General population mean score11,†
43.5
(SD: 8.3)ULTOMIRIS mean score2
42.8 at 5.6 years
(SD: 9.8)NOW APPROVED for the treatment of adult patients with anti-aquaporin-4 (AQP4) antibody-positive neuromyelitis optica spectrum disorder (NMOSD). 1
This information is intended for US healthcare professionals. If you are a healthcare professional, click OK to continue.
ULTOMIRIS was studied in the largest phase 3 PNH clinical trial program to date.1,2
*ULTOMIRIS was evaluated in two phase 3, open-label, randomized, active-controlled, multicenter, noninferiority studies that included a broad study population of clinically diverse adult patients (≥18 years) with PNH. See 301 Study Design (complement inhibitor–naïve) and 302 Study Design (clinically stable on eculizumab).1
PNH=paroxysmal nocturnal hemoglobinuria.
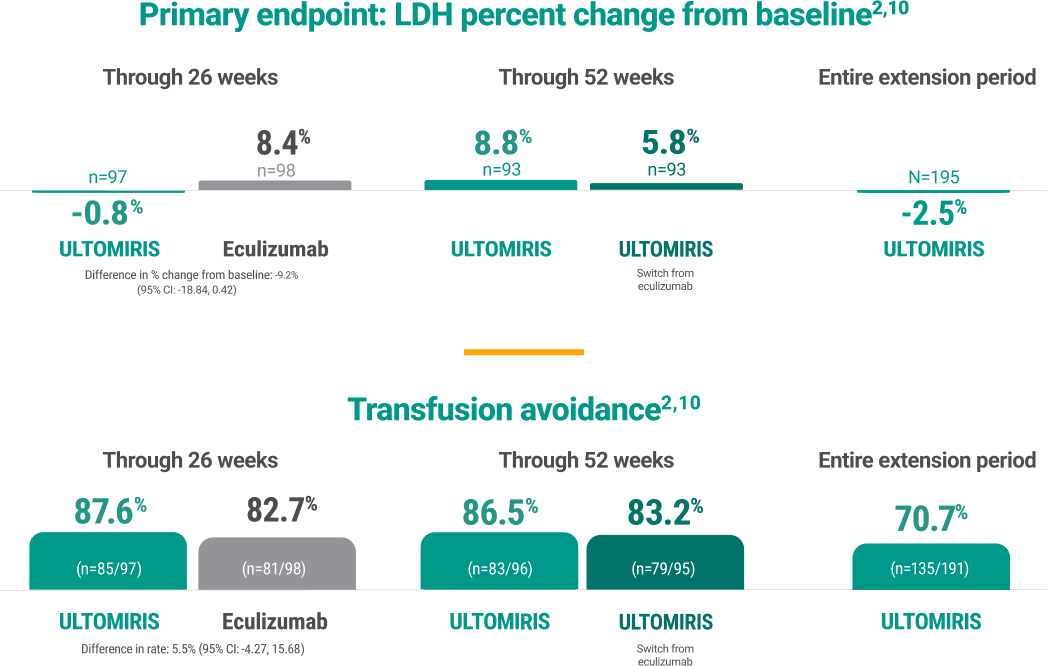
Noninferiority achieved
ULTOMIRIS was noninferior to eculizumab across all endpoints during the randomized treatment period (Week 0 to Week 26)1,4,5
Durable treatment response
Efficacy was maintained through the 5-year extension period, during which all patients received ULTOMIRIS.2

Treatment response continued throughout the 5-year study.1-4
LDH=lactate dehydrogenase.
LDH is an established biomarker of intravascular hemolysis and PNH disease activity1,6-8
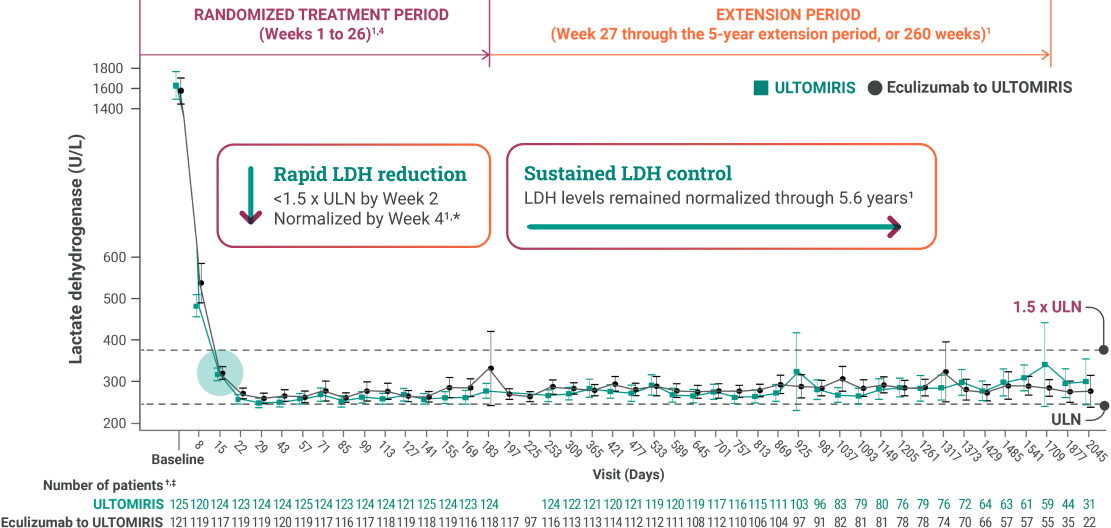
ULTOMIRIS helped reduce patients’ LDH to lower their risk of breakthrough IVH.5,§
*In patients receiving ULTOMIRIS, LDH levels rapidly fell below 1.5 x ULN by Week 2, normalized by Week 4, and were maintained below 1.5 x ULN through 5.6 years.1,2
†Number of patients may be lower than number enrolled at time point because of exclusion of samples having serum potassium ≥6 mmol/L and LDH ≥2 x ULN, missing samples (because of site error or for any other reason), or patient discontinuations during the extension.3
‡LDH levels were not measured for patients in the ULTOMIRIS group on Days 197 and 225.2
§Breakthrough IVH was defined as at least 1 new or worsening symptom or sign of IVH in the presence of elevated LDH ≥2 x ULN, after prior LDH reduction to <1.5 x ULN on therapy.2
IVH=intravascular hemolysis; LDH=lactate dehydrogenase; PNH=paroxysmal nocturnal hemoglobinuria; U/L=units per liter; ULN=upper limit of normal.
ULTOMIRIS helped reduce patients’ LDH to lower their risk of breakthrough IVH.5,§
ULTOMIRIS improved and maintained FACIT-Fatigue scores2,3,*

FACIT-Fatigue scores can range from 0 to 52—higher scores indicate less fatigue.11
43.5
(SD: 8.3)42.8 at 5.6 years
(SD: 9.8)*There was no observable difference in fatigue between ULTOMIRIS and eculizumab after 26 weeks of treatment compared to baseline as measured by the FACIT-Fatigue instrument. Patient-reported fatigue may be an underestimation or overestimation because patients were not blinded to treatment assignment.1
†Mean FACIT-Fatigue score for the general population was determined through assessment of 2,426 adults (n=1,074 males; n=1,352 females; mean age [SD]: 49.8 [17.4] years) in Germany between March 2015 and May 2015.11
FACIT=Functional Assessment of Chronic Illness Therapy; LS=least squares; SD=standard deviation.
Established to help stabilize hemoglobin (Hgb) levels in the majority of patients, including those with bone marrow failure throughout the study.

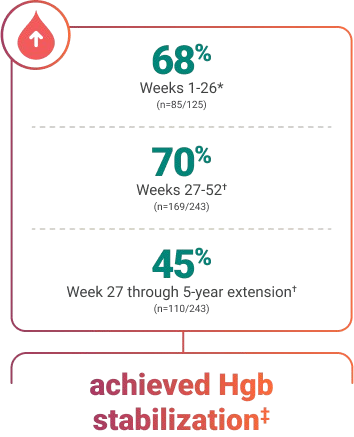
*Full analysis set (all patients who received ≥1 dose of ULTOMIRIS and had ≥1 efficacy assessment after the first infusion).3
†Extension set (all patients who entered the extension period).3
‡Defined as avoidance of a ≥2 g/dL decrease in Hgb level from baseline in the absence of a transfusion.2
96%
of patients did not experience
major adverse vascular events
through 5+ years2
(n=233/244)
Weeks 1-26: 1.6% (n=2/125); Weeks 27-52: 0.4% (n=1/243); entire study period: 4.5% (n=11/244)
Major adverse vascular events reported through the end of study were peripheral arterial thrombosis, coronary artery disease, cerebrovascular accident, angina unstable, deep vein thrombosis, acute myocardial infarction, pulmonary embolism, and cerebral venous thrombosis.2
86%
of patients did not experience
breakthrough IVH*
through the
5-year extension period2
(n=209/243)
Weeks 1-26: 4.0% (n=5/125); Weeks 27-52: 2.5% (n=6/243); entire extension period: 14.0% (n=34/243)
Weeks 1-26: 1.6% (n=2/125); Weeks 27-52: 0.4% (n=1/243); entire study period: 4.5% (n=11/244)
Major adverse vascular events reported through the end of study were peripheral arterial thrombosis, coronary artery disease, cerebrovascular accident, angina unstable, deep vein thrombosis, acute myocardial infarction, pulmonary embolism, and cerebral venous thrombosis.2
Weeks 1-26: 4.0% (n=5/125); Weeks 27-52: 2.5% (n=6/243); entire extension period: 14.0% (n=34/243)
*Breakthrough IVH was defined as at least 1 new or worsening symptom or sign of IVH in the presence of elevated LDH ≥2 x ULN, after prior LDH reduction to <1.5 x ULN on therapy.2
IVH=intravascular hemolysis; LDH=lactate dehydrogenase; MAVE=major adverse vascular event; PNH=paroxysmal nocturnal hemoglobinuria; ULN=upper limit of normal.
The phase 3, open-label, randomized, active-controlled, multicenter, noninferiority study included a broad study population of complement inhibitor–naïve, clinically diverse adult patients (≥18 years) with PNH1,2,4,9,*,† The objective of this study was to assess the noninferiority of ULTOMIRIS vs eculizumab in adult patients with PNH naïve to complement inhibitor therapy.4

Similar disease characteristics were observed in both study arms. Median pretreatment LDH levels were approximately 6 x ULN (246 U/L). Patients received a median of 6 units of transfused packed RBCs or whole blood in the 12 months preceding study enrollment.1,4
*Population included male and female patients ≥18 years of age in 25 countries with diagnosis of PNH confirmed by red and white blood cell (granulocyte or monocyte) clone sizes of ≥5%.2,3
†98% of patients had a documented PNH-associated condition diagnosed prior to enrollment in the trial: anemia (85%), hemoglobinuria (63%), history of aplastic anemia (32%), history of renal failure (12%), myelodysplastic syndrome (5%), pregnancy complications (3%), and other (16%).1
LDH=lactate dehydrogenase; PNH=paroxysmal nocturnal hemoglobinuria; RBC=red blood cell; U/L=units per liter; ULN=upper limit of normal.
| Baseline characteristics1 |
ULTOMIRIS (n=125) |
Eculizumab (n=121) |
| Pretreatment LDH levels, median U/L (min, max) |
1513.5 (378.0, 3759.5) |
1445.0 (423.5, 3139.5) |
| Transfusion units in preceding 12 months, median (min, max) | 6.0 (1, 44) | 6.0 (1, 32) |
| Antithrombotic agent use in preceding 28 days, n (%) | 22 (17.6) | 22 (18.2) |
| Concomitant anticoagulant treatment, n (%) | 23 (18.4) | 28 (23.1) |
| History of MAVEs, n (%) | 17 (13.6) | 25 (20.7) |
| History of thrombosis | 17 (13.6) | 20 (16.5) |
aplastic anemia
myelodysplastic syndrome
LDH=lactate dehydrogenase; MAVE=major adverse vascular event; RBC=red blood cell; U/L=units per liter.
*Defined as the patients who did not receive a transfusion and did not meet the protocol-specified guidelines for transfusion from baseline to Day 183.1
†Scores can range from 0 to 52, with higher scores indicating less fatigue.2
‡Defined as at least 1 new or worsening symptom or sign of intravascular hemolysis in the presence of elevated LDH ≥2 x ULN, after prior LDH reduction to <1.5 x ULN on therapy.2
§MAVEs included both thromboembolisms (thrombophlebitis/deep vein thrombosis, renal vein thrombosis, renal arterial thrombosis, mesenteric/visceral vein thrombosis or infarction, mesenteric/visceral arterial thrombosis or infarction, hepatic/portal vein thrombosis [Budd-Chiari syndrome], dermal thrombosis, acute peripheral vascular occlusion, cerebral arterial occlusion/cerebrovascular accident, cerebral venous occlusion, and pulmonary embolus) and nonthromboembolisms (myocardial infarction, transient ischemic attack, unstable angina, amputation [nontraumatic, nondiabetic], gangrene [nontraumatic, nondiabetic], and specified if other).2
||Defined as avoidance of a ≥2 g/dL decrease in hemoglobin level from baseline in the absence of transfusion.2
FACIT=Functional Assessment of Chronic Illness Therapy; IVH=intravascular hemolysis; LDH=lactate dehydrogenase; ULN=upper limit of normal.
Noninferiority achieved
ULTOMIRIS was noninferior to eculizumab across all endpoints during the randomized treatment period (Week 0 to Week 26)1,2
Durable treatment response
Efficacy was maintained through the 4-year extension period, during which all patients received ULTOMIRIS.2
*The extension period was up to 4 years for sites in Canada, France, and the Netherlands. For sites in Japan, the extension period was until the commercial product was delivered to the study site (no more than 3 years of study treatment).2
LDH=lactate dehydrogenase.

LDH=lactate dehydrogenase; U/L=units per liter; ULN=upper limit of normal.
ULTOMIRIS maintained FACIT-Fatigue scores through the 4-year extension period2,*
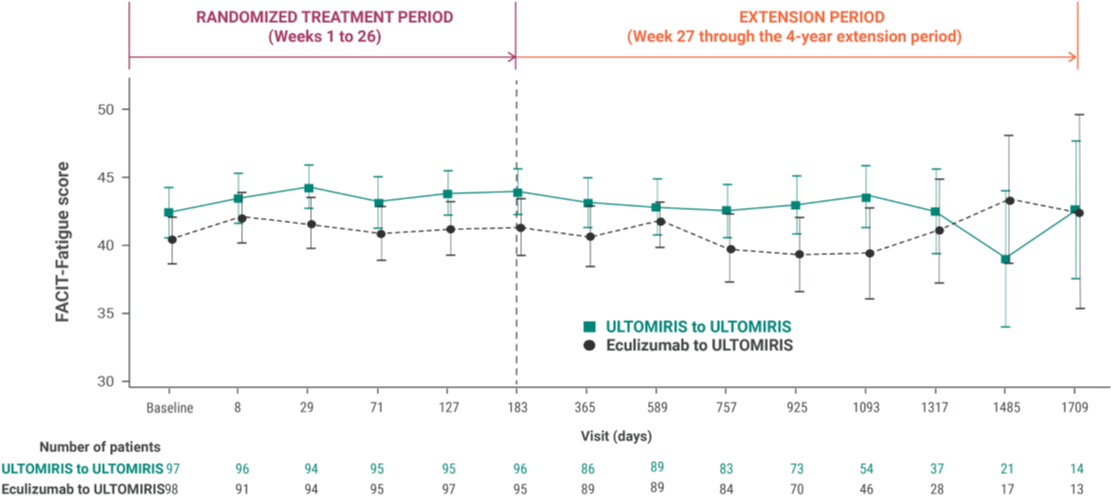
FACIT-Fatigue scores can range from 0 to 52—higher scores indicate less fatigue.11
43.5
42.0 at 3.6 years
*There was no observable difference in fatigue between ULTOMIRIS and eculizumab after 26 weeks of treatment compared to baseline as measured by the FACIT-Fatigue instrument. Patient-reported fatigue may be an underestimation or overestimation because patients were not blinded to treatment assignment.1
†Mean FACIT-Fatigue score for the general population was determined through assessment of 2,426 adults (n=1,074 males; n=1,352 females; mean age [SD]: 49.8 [17.4] years) in Germany between March 2015 and May 2015.11
FACIT=Functional Assessment of Chronic Illness Therapy; SD=standard deviation.
Established to help stabilize hemoglobin (Hgb) levels in the majority of patients, including those with bone marrow failure throughout the study.


*Full analysis set (all patients who received ≥1 dose of ULTOMIRIS and had ≥1 efficacy assessment after the first infusion).2
†Defined as avoidance of a ≥2 g/dL decrease in Hgb level from baseline in the absence of a transfusion.2
98%
of patients did not experience
major adverse vascular events
through the
4-year extension period2
(n=189/192)
92%
of patients did not experience
breakthrough IVH*
through the
4-year extension period2
(n=176/191)
Weeks 1-26: 0.0% (n=0/97); Weeks 27-52: 1.0% (n=2/191); entire study period: 1.6% (n=3/192)
Major adverse vascular events reported through the end of study were deep vein thrombosis, thrombophlebitis, and cerebral infarction.2,10
Weeks 1-26: 0.0% (n=0/97); Weeks 27-52: 2.0% (n=4/191); entire study period: 7.8% (n=15/191)
*Breakthrough IVH was defined as at least 1 new or worsening symptom or sign of intravascular hemolysis in the presence of elevated LDH ≥2 x ULN, after prior LDH reduction to <1.5 x ULN on therapy.10
IVH=intravascular hemolysis; LDH=lactate dehydrogenase; MAVE=major adverse vascular event; PNH=paroxysmal nocturnal hemoglobinuria; ULN=upper limit of normal.
The phase 3, open-label, randomized, active-controlled, multicenter, noninferiority study included a broad study population of clinically diverse adult patients (≥18 years) with PNH who were clinically stable on eculizumab for ≥6 months.1,2,*,† The objective of this study was to assess the noninferiority of ULTOMIRIS vs eculizumab in clinically stable PNH patients after previous eculizumab therapy.1,2.5

Similar disease characteristics were observed in both study arms. Patients received a median of 6 units of transfused packed RBCs or whole blood in the 12 months preceding study enrollment.1
*Population included male and female patients ≥18 years of age in 11 countries with diagnosis of PNH confirmed by red and white blood cell (granulocyte or monocyte) clone sizes of ≥5%.2
†95% of patients had a documented PNH-associated condition diagnosed prior to enrollment in the trial: anemia (67%), hematuria or hemoglobinuria (49%), history of aplastic anemia (37%), history of renal failure (9%), myelodysplastic syndrome (5%), pregnancy complications (7%), and other (15%).2
PNH=paroxysmal nocturnal hemoglobinuria; RBC=red blood cell.
| Baseline characteristics1 |
ULTOMIRIS (n=97) |
Eculizumab (n=98) |
| Pretreatment LDH levels, median U/L (min, max) |
224.0 (135.0, 383.5) |
234.0 (100.0, 365.5) |
| Transfusion units in preceding 12 months, median (min, max) | 4.0 (1, 32) | 2.5 (2, 15) |
| Antithrombotic agent use in preceding 28 days, n (%) | 20 (20.6) | 13 (13.3) |
| Concomitant anticoagulant treatment, n (%) | 22 (22.7) | 16 (16.3) |
| History of MAVEs, n (%) | 28 (28.9) | 22 (22.4) |
| History of thrombosis | 27 (27.8) | 21 (21.4) |
aplastic anemia
myelodysplastic syndrome
LDH=lactate dehydrogenase; MAVE=major adverse vascular event; U/L=units per liter.
LDH percent change from baseline
*Breakthrough hemolysis was defined as at least 1 new or worsening symptom or sign of intravascular hemolysis (fatigue, hemoglobinuria, abdominal pain, shortness of breath [dyspnea], anemia [hemoglobin <10 g/dL], MAVE [including thrombosis], dysphagia, or erectile dysfunction) in the presence of elevated LDH ≥2 x ULN after prior reduction of LDH to <1.5 x ULN on treatment.2,10
†FACIT-Fatigue scores can range from 0 to 52, with higher scores indicating less fatigue.11
‡Transfusion avoidance was defined as the proportion of patients who remained transfusion-free and did not require a transfusion per protocol-specified guidelines.2
§Stabilized hemoglobin was defined as avoidance of a ≥2 g/dL decrease in hemoglobin level from baseline in the absence of transfusion.2
FACIT=Functional Assessment of Chronic Illness Therapy; LDH=lactate dehydrogenase; MAVE=major adverse vascular event; ULN=upper limit of normal.
ULTOMIRIS has weight-based dosing with 6 or 7 infusions per year.1,*
*Starting 2 weeks after the intravenous loading dose, maintenance doses are infused intravenously every 8 weeks for adult patients and every 4 or 8 weeks for pediatric patients (depending on body weight).1
Find out how to get access to all of the tools necessary to get your patients started on ULTOMIRIS, including how to place an order.
References:
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
ULTOMIRIS, a complement inhibitor, increases the risk of serious infections caused by Neisseria meningitidis [see Warnings and Precautions (5.1)] Life-threatening and fatal meningococcal infections have occurred in patients treated with complement inhibitors. These infections may become rapidly life-threatening or fatal if not recognized and treated early.
Because of the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called ULTOMIRIS and SOLIRIS REMS [see Warnings and Precautions (5.2)].
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
Serious Meningococcal Infections
ULTOMIRIS, a complement inhibitor, increases a patient’s susceptibility to serious, life-threatening, or fatal infections caused by meningococcal bacteria (septicemia and/or meningitis) in any serogroup, including non-groupable strains. Life-threatening and fatal meningococcal infections have occurred in both vaccinated and unvaccinated patients treated with complement inhibitors.
Revaccinate patients in accordance with ACIP recommendations considering the duration of ULTOMIRIS therapy. Note that ACIP recommends an administration schedule in patients receiving complement inhibitors that differs from the administration schedule in the vaccine prescribing information. If urgent ULTOMIRIS therapy is indicated in a patient who is not up to date with meningococcal vaccines according to ACIP recommendations, provide antibacterial drug prophylaxis and administer meningococcal vaccines as soon as possible. Various durations and regimens of antibacterial drug prophylaxis have been considered, but the optimal durations and drug regimens for prophylaxis and their efficacy have not been studied in unvaccinated or vaccinated patients receiving complement inhibitors, including ULTOMIRIS. The benefits and risks of treatment with ULTOMIRIS, as well as those associated with antibacterial drug prophylaxis in unvaccinated or vaccinated patients, must be considered against the known risks for serious infections caused by Neisseria meningitidis.
Vaccination does not eliminate the risk of serious meningococcal infections, despite development of antibodies following vaccination.
Closely monitor patients for early signs and symptoms of meningococcal infection and evaluate patients immediately if infection is suspected. Inform patients of these signs and symptoms and instruct patients to seek immediate medical care if they occur. Promptly treat known infections. Meningococcal infection may become rapidly life-threatening or fatal if not recognized and treated early. Consider interruption of ULTOMIRIS in patients who are undergoing treatment for serious meningococcal infection depending on the risks of interrupting treatment in the disease being treated.
ULTOMIRIS and SOLIRIS REMS
Due to the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program called ULTOMIRIS and SOLIRIS REMS.
Prescribers must enroll in the REMS, counsel patients about the risk of serious meningococcal infection, provide patients with the REMS educational materials, assess patient vaccination status for meningococcal vaccines (against serogroups A, C, W, Y, and B) and vaccinate if needed according to current ACIP recommendations two weeks prior to the first dose of ULTOMIRIS. Antibacterial drug prophylaxis must be prescribed if treatment must be started urgently, and the patient is not up to date with both meningococcal vaccines according to current ACIP recommendations at least two weeks prior to the first dose of ULTOMIRIS. Patients must receive counseling about the need to receive meningococcal vaccines and to take antibiotics as directed, signs and symptoms of meningococcal infection, and be instructed to carry the Patient Safety Card at all times during and for 8 months following ULTOMIRIS treatment.
Further information is available at www.UltSolREMS.com or 1-888-765-4747.
Other Infections
Serious infections with Neisseria species (other than Neisseria meningitidis), including disseminated gonococcal infections, have been reported.
ULTOMIRIS blocks terminal complement activation; therefore, patients may have increased susceptibility to infections, especially with encapsulated bacteria, such as infections caused by Neisseria meningitidis but also Streptococcus pneumoniae, Haemophilus influenzae, and to a lesser extent, Neisseria gonorrhoeae. Children treated with ULTOMIRIS may be at increased risk of developing serious infections due to Streptococcus pneumoniae and Haemophilus influenzae type b (Hib). Administer vaccinations for the prevention of Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) infections according to ACIP recommendations. Patients receiving ULTOMIRIS are at increased risk for infections due to these organisms, even if they develop antibodies following vaccination.
Monitoring Disease Manifestations after ULTOMIRIS Discontinuation
Treatment Discontinuation for PNH
After discontinuing treatment with ULTOMIRIS, closely monitor for signs and symptoms of hemolysis, identified by elevated LDH along with sudden decrease in PNH clone size or hemoglobin, or re-appearance of symptoms such as fatigue, hemoglobinuria, abdominal pain, shortness of breath (dyspnea), major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction. Monitor any patient who discontinues ULTOMIRIS for at least 16 weeks to detect hemolysis and other reactions. If signs and symptoms of hemolysis occur after discontinuation, including elevated LDH, consider restarting treatment with ULTOMIRIS.
Treatment Discontinuation for aHUS
ULTOMIRIS treatment of aHUS should be a minimum duration of 6 months. Due to heterogeneous nature of aHUS events and patient-specific risk factors, treatment duration beyond the initial 6 months should be individualized. There are no specific data on ULTOMIRIS discontinuation. After discontinuing treatment with ULTOMIRIS, patients should be monitored for clinical symptoms and laboratory signs of TMA complications for at least 12 months. TMA complications post-discontinuation can be identified if any of the following is observed: Clinical symptoms of TMA include changes in mental status, seizures, angina, dyspnea, thrombosis or increasing blood pressure. In addition, at least two of the following laboratory signs observed concurrently and results should be confirmed by a second measurement 28 days apart with no interruption: a decrease in platelet count of 25% or more as compared to either baseline or to peak platelet count during ULTOMIRIS treatment; an increase in serum creatinine of 25% or more as compared to baseline or to nadir during ULTOMIRIS treatment; or, an increase in serum LDH of 25% or more as compared to baseline or to nadir during ULTOMIRIS treatment. If TMA complications occur after discontinuation, consider reinitiation of ULTOMIRIS treatment or appropriate organ-specific supportive measures.
Thromboembolic Event Management
The effect of withdrawal of anticoagulant therapy during treatment with ULTOMIRIS has not been established. Treatment should not alter anticoagulant management.
Infusion-Related Reactions
Administration of ULTOMIRIS may result in systemic infusion-related reactions, including anaphylaxis and hypersensitivity reactions. In clinical trials, infusion-related reactions occurred in approximately 1 to 7% of patients, including lower back pain, abdominal pain, muscle spasms, drop or elevation in blood pressure, rigors, limb discomfort, drug hypersensitivity (allergic reaction), and dysgeusia (bad taste). These reactions did not require discontinuation of ULTOMIRIS. If signs of cardiovascular instability or respiratory compromise occur, interrupt ULTOMIRIS and institute appropriate supportive measures.
ADVERSE REACTIONS
Adverse Reactions for PNH
Adverse reactions reported in ≥10% or more of patients with PNH were upper respiratory tract infection and headache. Serious adverse reactions were reported in 15 (6.8%) patients receiving ULTOMIRIS. The serious adverse reactions in patients treated with ULTOMIRIS included hyperthermia and pyrexia. No serious adverse reaction was reported in more than 1 patient treated with ULTOMIRIS. One fatal case of sepsis was identified in a patient treated with ULTOMIRIS. In clinical studies, clinically relevant adverse reactions in 1% of adult patients include infusion-related reactions.
Adverse reactions reported in ≥10% of pediatric patients treated with ULTOMIRIS who were treatment-naïve vs. Eculizumab-experienced were anemia (20% vs. 25%), abdominal pain (0% vs. 38%), constipation (0% vs. 25%), pyrexia (20% vs. 13%), upper respiratory tract infection (20% vs. 75%), pain in extremity (0% vs. 25%), and headache (20% vs. 25%).
Adverse Reactions for aHUS
Most common adverse reactions in patients with aHUS (incidence ≥20%) were upper respiratory tract infection, diarrhea, nausea, vomiting, headache, hypertension and pyrexia. Serious adverse reactions were reported in 42 (57%) patients with aHUS receiving ULTOMIRIS. The most frequent serious adverse reactions reported in more than 2 patients (2.7%) treated with ULTOMIRIS were hypertension, pneumonia and abdominal pain.
Adverse reactions reported in ≥20% of pediatric patients treated with ULTOMIRIS were diarrhea, constipation, vomiting, pyrexia, upper respiratory tract infection, decreased vitamin D, headache, cough, rash, and hypertension.
Adverse Reactions for gMG
Most common adverse reactions in adult patients with gMG (incidence ≥10%) were diarrhea and upper respiratory tract infection. Serious adverse reactions were reported in 20 (23%) of patients treated with ULTOMIRIS and in 14 (16%) patients receiving placebo. The most frequent serious adverse reactions were infections reported in at least 8 (9%) patients treated with ULTOMIRIS and in 4 (4%) patients treated with placebo. Of these infections, one fatal case of COVID-19 pneumonia was identified in a patient treated with ULTOMIRIS and one case of infection led to discontinuation of ULTOMIRIS.
Adverse Reactions for NMOSD
Most common adverse reactions in adult patients with NMOSD (incidence ≥10%) were COVID-19, headache, back pain, arthralgia, and urinary tract infection. Serious adverse reactions were reported in 8 (13.8%) patients with NMOSD receiving ULTOMIRIS.
DRUG INTERACTIONS
Plasma Exchange, Plasmapheresis, and Intravenous Immunoglobulins
Concomitant use of ULTOMIRIS with plasma exchange (PE), plasmapheresis (PP), or intravenous immunoglobulin (IVIg) treatment can reduce serum ravulizumab concentrations and requires a supplemental dose of ULTOMIRIS.
Neonatal Fc Receptor Blockers
Concomitant use of ULTOMIRIS with neonatal Fc receptor (FcRn) blockers (e.g., efgartigimod) may lower systemic exposures and reduce effectiveness of ULTOMIRIS. Closely monitor for reduced effectiveness of ULTOMIRIS.
USE IN SPECIFIC POPULATIONS
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ULTOMIRIS during pregnancy. Healthcare providers and patients may call 1-833-793-0563 or go to www.UltomirisPregnancyStudy.com to enroll in or to obtain information about the registry.
To report SUSPECTED ADVERSE REACTIONS, contact Alexion Pharmaceuticals, Inc. at 1-844-259-6783 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
INDICATIONS
Paroxysmal Nocturnal Hemoglobinuria (PNH)
ULTOMIRIS is indicated for the treatment of adult and pediatric patients one month of age and older with paroxysmal nocturnal hemoglobinuria (PNH).
Atypical Hemolytic Uremic Syndrome (aHUS)
ULTOMIRIS is indicated for the treatment of adult and pediatric patients one month of age and older with atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy (TMA).
Limitation of Use:
ULTOMIRIS is not indicated for the treatment of patients with Shiga toxin E. coli related hemolytic uremic syndrome (STEC-HUS).
Generalized Myasthenia Gravis (gMG)
ULTOMIRIS is indicated for the treatment of adult patients with generalized myasthenia gravis (gMG) who are anti-acetylcholine receptor (AChR) antibody-positive.
Neuromyelitis Optica Spectrum Disorder (NMOSD)
ULTOMIRIS is indicated for the treatment of adult patients with neuromyelitis optica spectrum disorder (NMOSD) who are anti-aquaporin-4 (AQP4) antibody positive.
Please see full Prescribing Information for ULTOMIRIS, including Boxed WARNING regarding serious and life-threatening or fatal meningococcal infections.
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
ULTOMIRIS, a complement inhibitor, increases the risk of serious infections caused by Neisseria meningitidis [see Warnings and Precautions (5.1)] Life-threatening and fatal meningococcal infections have occurred in patients treated with complement inhibitors. These infections may become rapidly life-threatening or fatal if not recognized and treated early.
Because of the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called ULTOMIRIS and SOLIRIS REMS [see Warnings and Precautions (5.2)].
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
Serious Meningococcal Infections
ULTOMIRIS, a complement inhibitor, increases a patient’s susceptibility to serious, life-threatening, or fatal infections caused by meningococcal bacteria (septicemia and/or meningitis) in any serogroup, including non-groupable strains. Life-threatening and fatal meningococcal infections have occurred in both vaccinated and unvaccinated patients treated with complement inhibitors.
Revaccinate patients in accordance with ACIP recommendations considering the duration of ULTOMIRIS therapy. Note that ACIP recommends an administration schedule in patients receiving complement inhibitors that differs from the administration schedule in the vaccine prescribing information. If urgent ULTOMIRIS therapy is indicated in a patient who is not up to date with meningococcal vaccines according to ACIP recommendations, provide antibacterial drug prophylaxis and administer meningococcal vaccines as soon as possible. Various durations and regimens of antibacterial drug prophylaxis have been considered, but the optimal durations and drug regimens for prophylaxis and their efficacy have not been studied in unvaccinated or vaccinated patients receiving complement inhibitors, including ULTOMIRIS. The benefits and risks of treatment with ULTOMIRIS, as well as those associated with antibacterial drug prophylaxis in unvaccinated or vaccinated patients, must be considered against the known risks for serious infections caused by Neisseria meningitidis.
Vaccination does not eliminate the risk of serious meningococcal infections, despite development of antibodies following vaccination.
Closely monitor patients for early signs and symptoms of meningococcal infection and evaluate patients immediately if infection is suspected. Inform patients of these signs and symptoms and instruct patients to seek immediate medical care if they occur. Promptly treat known infections. Meningococcal infection may become rapidly life-threatening or fatal if not recognized and treated early. Consider interruption of ULTOMIRIS in patients who are undergoing treatment for serious meningococcal infection depending on the risks of interrupting treatment in the disease being treated.
ULTOMIRIS and SOLIRIS REMS
Due to the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program called ULTOMIRIS and SOLIRIS REMS.
Prescribers must enroll in the REMS, counsel patients about the risk of serious meningococcal infection, provide patients with the REMS educational materials, assess patient vaccination status for meningococcal vaccines (against serogroups A, C, W, Y, and B) and vaccinate if needed according to current ACIP recommendations two weeks prior to the first dose of ULTOMIRIS. Antibacterial drug prophylaxis must be prescribed if treatment must be started urgently, and the patient is not up to date with both meningococcal vaccines according to current ACIP recommendations at least two weeks prior to the first dose of ULTOMIRIS. Patients must receive counseling about the need to receive meningococcal vaccines and to take antibiotics as directed, signs and symptoms of meningococcal infection, and be instructed to carry the Patient Safety Card at all times during and for 8 months following ULTOMIRIS treatment.
Further information is available at www.UltSolREMS.com or 1-888-765-4747.
Other Infections
Serious infections with Neisseria species (other than Neisseria meningitidis), including disseminated gonococcal infections, have been reported.
ULTOMIRIS blocks terminal complement activation; therefore, patients may have increased susceptibility to infections, especially with encapsulated bacteria, such as infections caused by Neisseria meningitidis but also Streptococcus pneumoniae, Haemophilus influenzae, and to a lesser extent, Neisseria gonorrhoeae. Children treated with ULTOMIRIS may be at increased risk of developing serious infections due to Streptococcus pneumoniae and Haemophilus influenzae type b (Hib). Administer vaccinations for the prevention of Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) infections according to ACIP recommendations. Patients receiving ULTOMIRIS are at increased risk for infections due to these organisms, even if they develop antibodies following vaccination.
Monitoring Disease Manifestations after ULTOMIRIS Discontinuation
After discontinuing treatment with ULTOMIRIS, closely monitor for signs and symptoms of hemolysis, identified by elevated LDH along with sudden decrease in PNH clone size or hemoglobin, or re-appearance of symptoms such as fatigue, hemoglobinuria, abdominal pain, shortness of breath (dyspnea), major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction. Monitor any patient who discontinues ULTOMIRIS for at least 16 weeks to detect hemolysis and other reactions. If signs and symptoms of hemolysis occur after discontinuation, including elevated LDH, consider restarting treatment with ULTOMIRIS.
Thromboembolic Event Management
The effect of withdrawal of anticoagulant therapy during treatment with ULTOMIRIS has not been established. Treatment should not alter anticoagulant management.
Infusion-Related Reactions
Administration of ULTOMIRIS may result in systemic infusion-related reactions, including anaphylaxis and hypersensitivity reactions. In clinical trials, infusion-related reactions occurred in approximately 1 to 7% of patients, including lower back pain, abdominal pain, muscle spasms, drop or elevation in blood pressure, rigors, limb discomfort, drug hypersensitivity (allergic reaction), and dysgeusia (bad taste). These reactions did not require discontinuation of ULTOMIRIS. If signs of cardiovascular instability or respiratory compromise occur, interrupt ULTOMIRIS and institute appropriate supportive measures.
ADVERSE REACTIONS
Adverse reactions reported in ≥10% or more of patients with PNH were upper respiratory tract infection and headache. Serious adverse reactions were reported in 15 (6.8%) patients receiving ULTOMIRIS. The serious adverse reactions in patients treated with ULTOMIRIS included hyperthermia and pyrexia. No serious adverse reaction was reported in more than 1 patient treated with ULTOMIRIS. One fatal case of sepsis was identified in a patient treated with ULTOMIRIS. In clinical studies, clinically relevant adverse reactions in 1% of adult patients include infusion-related reactions.
Adverse reactions reported in ≥10% of pediatric patients treated with ULTOMIRIS who were treatment-naïve vs. Eculizumab-experienced were anemia (20% vs. 25%), abdominal pain (0% vs. 38%), constipation (0% vs. 25%), pyrexia (20% vs. 13%), upper respiratory tract infection (20% vs. 75%), pain in extremity (0% vs. 25%), and headache (20% vs. 25%).
DRUG INTERACTIONS
Plasma Exchange, Plasmapheresis, and Intravenous Immunoglobulins
Concomitant use of ULTOMIRIS with plasma exchange (PE), plasmapheresis (PP), or intravenous immunoglobulin (IVIg) treatment can reduce serum ravulizumab concentrations and requires a supplemental dose of ULTOMIRIS.
Neonatal Fc Receptor Blockers
Concomitant use of ULTOMIRIS with neonatal Fc receptor (FcRn) blockers (e.g., efgartigimod) may lower systemic exposures and reduce effectiveness of ULTOMIRIS. Closely monitor for reduced effectiveness of ULTOMIRIS.
USE IN SPECIFIC POPULATIONS
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ULTOMIRIS during pregnancy. Healthcare providers and patients may call 1-833-793-0563 or go to www.UltomirisPregnancyStudy.com to enroll in or to obtain information about the registry.
INDICATION
ULTOMIRIS is indicated for the treatment of adult and pediatric patients one month of age and older with paroxysmal nocturnal hemoglobinuria (PNH).
To report SUSPECTED ADVERSE REACTIONS, contact Alexion Pharmaceuticals, Inc. at 1-844-259-6783 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Please see full Prescribing Information for ULTOMIRIS, including Boxed WARNING regarding serious and life-threatening or fatal meningococcal infections.
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
ULTOMIRIS, a complement inhibitor, increases the risk of serious infections caused by Neisseria meningitidis [see Warnings and Precautions (5.1)] Life-threatening and fatal meningococcal infections have occurred in patients treated with complement inhibitors. These infections may become rapidly life-threatening or fatal if not recognized and treated early.
Because of the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called ULTOMIRIS and SOLIRIS REMS [see Warnings and Precautions (5.2)].
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
Serious Meningococcal Infections
ULTOMIRIS, a complement inhibitor, increases a patient’s susceptibility to serious, life-threatening, or fatal infections caused by meningococcal bacteria (septicemia and/or meningitis) in any serogroup, including non-groupable strains. Life-threatening and fatal meningococcal infections have occurred in both vaccinated and unvaccinated patients treated with complement inhibitors.
Revaccinate patients in accordance with ACIP recommendations considering the duration of ULTOMIRIS therapy. Note that ACIP recommends an administration schedule in patients receiving complement inhibitors that differs from the administration schedule in the vaccine prescribing information. If urgent ULTOMIRIS therapy is indicated in a patient who is not up to date with meningococcal vaccines according to ACIP recommendations, provide antibacterial drug prophylaxis and administer meningococcal vaccines as soon as possible. Various durations and regimens of antibacterial drug prophylaxis have been considered, but the optimal durations and drug regimens for prophylaxis and their efficacy have not been studied in unvaccinated or vaccinated patients receiving complement inhibitors, including ULTOMIRIS. The benefits and risks of treatment with ULTOMIRIS, as well as those associated with antibacterial drug prophylaxis in unvaccinated or vaccinated patients, must be considered against the known risks for serious infections caused by Neisseria meningitidis.
Vaccination does not eliminate the risk of serious meningococcal infections, despite development of antibodies following vaccination.
Closely monitor patients for early signs and symptoms of meningococcal infection and evaluate patients immediately if infection is suspected. Inform patients of these signs and symptoms and instruct patients to seek immediate medical care if they occur. Promptly treat known infections. Meningococcal infection may become rapidly life-threatening or fatal if not recognized and treated early. Consider interruption of ULTOMIRIS in patients who are undergoing treatment for serious meningococcal infection depending on the risks of interrupting treatment in the disease being treated.
ULTOMIRIS and SOLIRIS REMS
Due to the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program called ULTOMIRIS and SOLIRIS REMS.
Prescribers must enroll in the REMS, counsel patients about the risk of serious meningococcal infection, provide patients with the REMS educational materials, assess patient vaccination status for meningococcal vaccines (against serogroups A, C, W, Y, and B) and vaccinate if needed according to current ACIP recommendations two weeks prior to the first dose of ULTOMIRIS. Antibacterial drug prophylaxis must be prescribed if treatment must be started urgently, and the patient is not up to date with both meningococcal vaccines according to current ACIP recommendations at least two weeks prior to the first dose of ULTOMIRIS. Patients must receive counseling about the need to receive meningococcal vaccines and to take antibiotics as directed, signs and symptoms of meningococcal infection, and be instructed to carry the Patient Safety Card at all times during and for 8 months following ULTOMIRIS treatment.
Further information is available at www.UltSolREMS.com or 1-888-765-4747.
Other Infections
Serious infections with Neisseria species (other than Neisseria meningitidis), including disseminated gonococcal infections, have been reported.
ULTOMIRIS blocks terminal complement activation; therefore, patients may have increased susceptibility to infections, especially with encapsulated bacteria, such as infections caused by Neisseria meningitidis but also Streptococcus pneumoniae, Haemophilus influenzae, and to a lesser extent, Neisseria gonorrhoeae. Children treated with ULTOMIRIS may be at increased risk of developing serious infections due to Streptococcus pneumoniae and Haemophilus influenzae type b (Hib). Administer vaccinations for the prevention of Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) infections according to ACIP recommendations. Patients receiving ULTOMIRIS are at increased risk for infections due to these organisms, even if they develop antibodies following vaccination.
Monitoring Disease Manifestations after ULTOMIRIS Discontinuation
ULTOMIRIS treatment of aHUS should be a minimum duration of 6 months. Due to heterogeneous nature of aHUS events and patient-specific risk factors, treatment duration beyond the initial 6 months should be individualized. There are no specific data on ULTOMIRIS discontinuation. After discontinuing treatment with ULTOMIRIS, patients should be monitored for clinical symptoms and laboratory signs of TMA complications for at least 12 months. TMA complications post-discontinuation can be identified if any of the following is observed: Clinical symptoms of TMA include changes in mental status, seizures, angina, dyspnea, thrombosis or increasing blood pressure. In addition, at least two of the following laboratory signs observed concurrently and results should be confirmed by a second measurement 28 days apart with no interruption: a decrease in platelet count of 25% or more as compared to either baseline or to peak platelet count during ULTOMIRIS treatment; an increase in serum creatinine of 25% or more as compared to baseline or to nadir during ULTOMIRIS treatment; or, an increase in serum LDH of 25% or more as compared to baseline or to nadir during ULTOMIRIS treatment. If TMA complications occur after discontinuation, consider reinitiation of ULTOMIRIS treatment or appropriate organ-specific supportive measures.
Thromboembolic Event Management
The effect of withdrawal of anticoagulant therapy during treatment with ULTOMIRIS has not been established. Treatment should not alter anticoagulant management.
Infusion-Related Reactions
Administration of ULTOMIRIS may result in systemic infusion-related reactions, including anaphylaxis and hypersensitivity reactions. In clinical trials, infusion-related reactions occurred in approximately 1 to 7% of patients, including lower back pain, abdominal pain, muscle spasms, drop or elevation in blood pressure, rigors, limb discomfort, drug hypersensitivity (allergic reaction), and dysgeusia (bad taste). These reactions did not require discontinuation of ULTOMIRIS. If signs of cardiovascular instability or respiratory compromise occur, interrupt ULTOMIRIS and institute appropriate supportive measures.
ADVERSE REACTIONS
Most common adverse reactions in patients with aHUS (incidence ≥20%) were upper respiratory tract infection, diarrhea, nausea, vomiting, headache, hypertension and pyrexia. Serious adverse reactions were reported in 42 (57%) patients with aHUS receiving ULTOMIRIS. The most frequent serious adverse reactions reported in more than 2 patients (2.7%) treated with ULTOMIRIS were hypertension, pneumonia and abdominal pain.
Adverse reactions reported in ≥20% of pediatric patients treated with ULTOMIRIS were diarrhea, constipation, vomiting, pyrexia, upper respiratory tract infection, decreased vitamin D, headache, cough, rash, and hypertension.
DRUG INTERACTIONS
Plasma Exchange, Plasmapheresis, and Intravenous Immunoglobulins
Concomitant use of ULTOMIRIS with plasma exchange (PE), plasmapheresis (PP), or intravenous immunoglobulin (IVIg) treatment can reduce serum ravulizumab concentrations and requires a supplemental dose of ULTOMIRIS.
Neonatal Fc Receptor Blockers
Concomitant use of ULTOMIRIS with neonatal Fc receptor (FcRn) blockers (e.g., efgartigimod) may lower systemic exposures and reduce effectiveness of ULTOMIRIS. Closely monitor for reduced effectiveness of ULTOMIRIS.
USE IN SPECIFIC POPULATIONS
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ULTOMIRIS during pregnancy. Healthcare providers and patients may call 1-833-793-0563 or go to www.UltomirisPregnancyStudy.com to enroll in or to obtain information about the registry.
INDICATION
ULTOMIRIS is indicated for the treatment of adult and pediatric patients one month of age and older with atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy (TMA).
Limitation of Use:
ULTOMIRIS is not indicated for the treatment of patients with Shiga toxin E. coli related hemolytic uremic syndrome (STEC‑HUS).
To report SUSPECTED ADVERSE REACTIONS, contact Alexion Pharmaceuticals, Inc. at 1-844-259-6783 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Please see full Prescribing Information for ULTOMIRIS, including Boxed WARNING regarding serious and life-threatening or fatal meningococcal infections.