ULTOMIRIS pediatric efficacy and safety
ULTOMIRIS, the first and only long-acting complement inhibitor for atypical-HUS, provided immediate, complete, and sustained complement inhibition in adult and pediatric patients 1 month of age and older.1
For treatment of atypical-HUS in adult and pediatric patients 1 month of age and older. Not indicated in STEC‑HUS.1
STEC-HUS=Shiga toxin–producing E coli hemolytic uremic syndrome.
Actor portrayal
aAs measured by free C5 serum concentration of <0.5 mcg/mL.1
bComplete TMA response was defined as normalization of hematological parameters (platelet count and LDH) and ≥25% improvement in serum creatinine from baseline during the 26-week initial evaluation period. Patients had to meet all criteria at 2 separate assessments obtained at least 4 weeks (28 days) apart and any measurement in between.1
C5=complement protein 5; LDH=lactate dehydrogenase; TMA=thrombotic microangiopathy.
Complete TMA response
Nearly 3 out of 4 pediatric patients (n=10/14) taking ULTOMIRIS achieved complete TMA response, which was maintained by all patients at Week 501,3,c
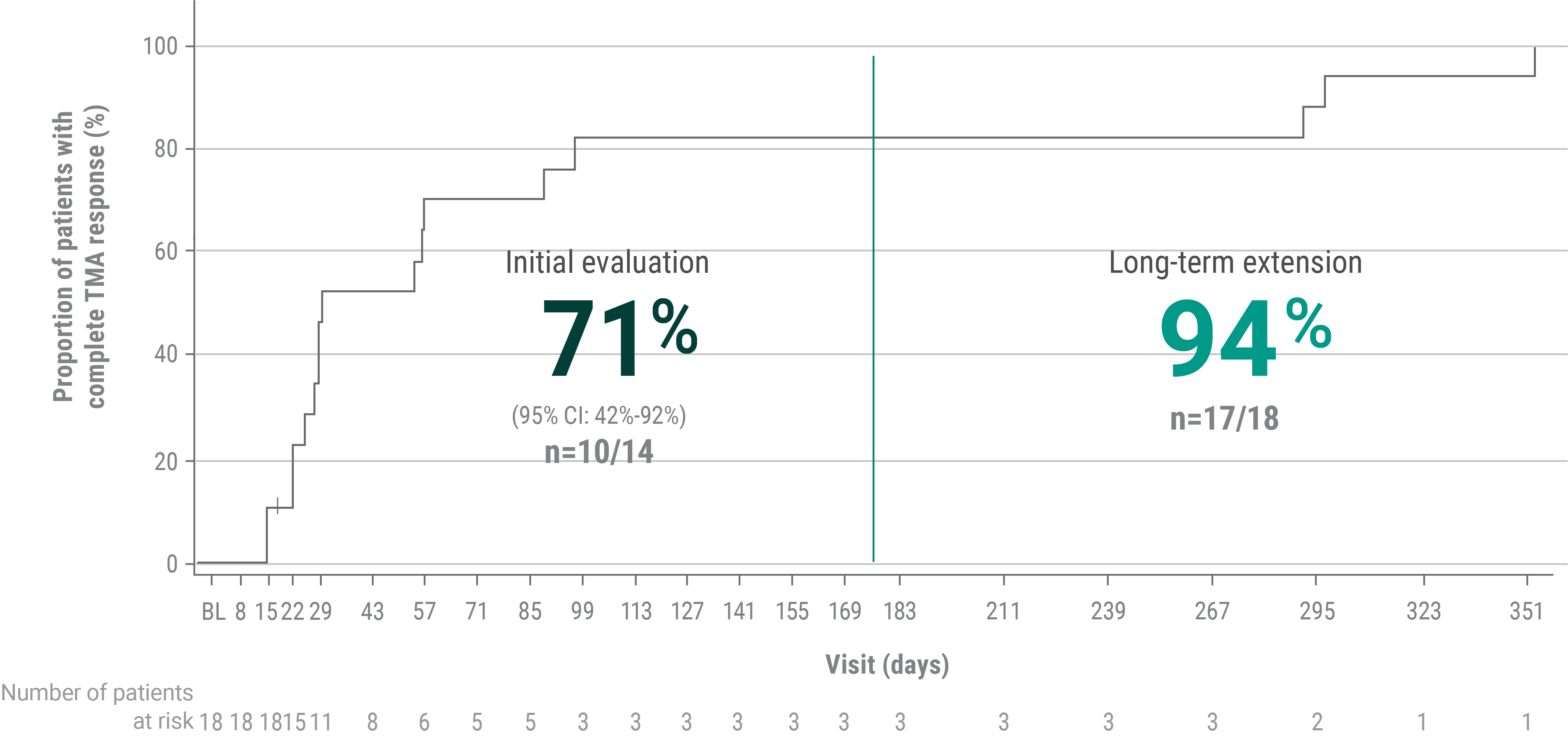
Median time to complete TMA response was 30.0 days (95% CI: 22.0, 88.0).3
Complete TMA response
Children
Median time to complete TMA response was 30.0 days (95% CI: 22.0, 88.0).3
The interim analysis at 26 weeks was performed with 14 pediatric patients with atypical-HUS. As the study was ongoing, additional patients were recruited, and the 50-week analysis included 18 pediatric patients.
cComplete TMA response was defined as normalization of hematological parameters (platelet count and LDH) and ≥25% improvement in serum creatinine from baseline during the 26-week initial evaluation period. Patients had to meet all criteria at 2 separate assessments obtained at least 4 weeks (28 days) apart and any measurement in between.1 Patients that did not have a response were censored at the date of last visit or study discontinuation at the time when the analysis was performed.2
BL=baseline; LDH=lactate dehydrogenase; TMA=thrombotic microangiopathy.
Components of complete TMA response1,3,d
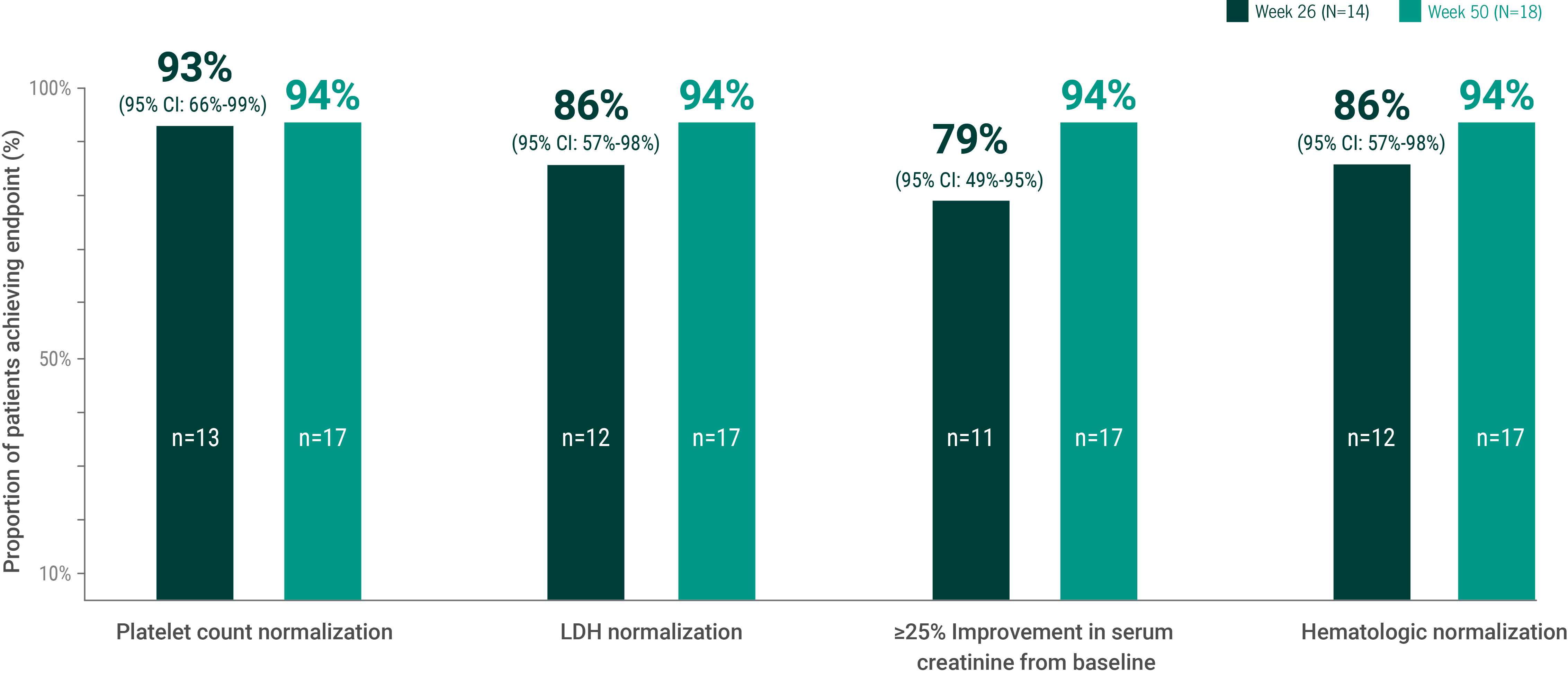
Platelet count normalization
Children
LDH normalization
Children
≥25% Improvement in serum creatinine from baseline
Children
Hematologic normalization
Children
dComplete TMA response during the 26-week initial evaluation period was summarized by number and proportion of complete TMA responders (with 2-sided 95% confidence interval). Statistical analysis was not performed on 50-week data.
LDH=lactate dehydrogenase; TMA=thrombotic microangiopathy.
eAs measured by free C5 serum concentration of <0.5 mcg/mL.1
C5=complement protein 5.
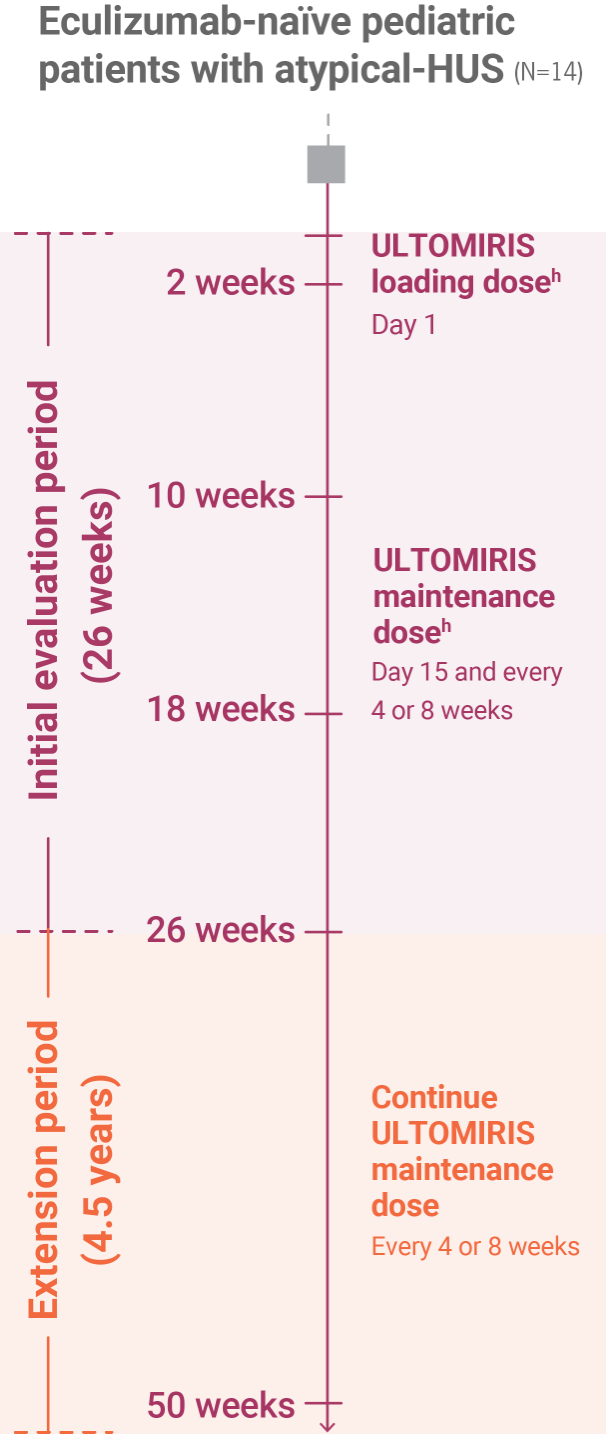
Study design
ULTOMIRIS was evaluated in a 26-week study in pediatric patients with atypical-HUS, followed by an ongoing extension study1,3
Pivotal study and extension study1,3

hULTOMIRIS weight-based dosing regimen.
Select inclusion criteria:
- Platelet count ≤150 x 109 cells/L
- Evidence of hemolysis (eg, elevated serum LDH, serum creatinine ≥97.5%, or required dialysis)
Select exclusion criteria:
- Patients with TMA due to ADAMTS13 deficiency, STEC‑HUS, and genetic defect in cobalamin C metabolism
Pivotal study
- The pivotal study was conducted in 16 pediatric patients. A total of 14 eculizumab-naïve patients with documented diagnosis of atypical-HUS were enrolled and included in the interim 26-week analysis.
Extension study
- Pediatric patients with atypical-HUS in the 26-week initial evaluation period could enter an ongoing 4.5-year extension period.
- Long-term extension data are from an interim analysis with 18 patients at Week 50.
ADAMTS13=a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13; LDH=lactate dehydrogenase; STEC-HUS=Shiga toxin–producing E coli hemolytic uremic syndrome; TMA=thrombotic microangiopathy.
Select baseline characteristics1,2
iPatients can have multiple races selected.
CKD=chronic kidney disease; eGFR=estimated glomerular filtration rate; LDH=lactate dehydrogenase; U/L=units per liter.
Study endpoints2
Primary endpoint
Composite endpoint of complete TMA response:
- Platelet count normalizationj
- Serum LDH normalizationj
- ≥25% improvement in serum creatinine from baseline
Patients had to meet all criteria for the primary endpoint of a complete TMA response at 2 separate assessments obtained at least 4 weeks (28 days) apart and any measurement in between.
jIncludes ≥150 x 109 cells/L for platelet count and ≤246 U/L for LDH.2
Select secondary endpoints
- Time to complete TMA response and complete TMA response status over time
- Dialysis requirement and CKD stage as evaluated by eGFR
- Hemoglobin response
- Change from baseline in quality of life
CKD=chronic kidney disease; eGFR=estimated glomerular filtration rate; LDH=lactate dehydrogenase; TMA=thrombotic microangiopathy; U/L=units per liter.
This way to adult efficacy data
Read the efficacy and safety data for ULTOMIRIS studied in adult patients with atypical-HUS.
Explore adult dataTools for the journey
Find additional resources and support information to help get your patients started on ULTOMIRIS.
View resources- ULTOMIRIS. Prescribing information. Alexion Pharmaceuticals, Inc.
- Data on file [ALXN1210-aHUS-312CSR].
- Ariceta G, et al. Kidney Int. 2021;100(1):225-237.
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
ULTOMIRIS, a complement inhibitor, increases the risk of serious infections caused by Neisseria meningitidis [see Warnings and Precautions (5.1)] Life-threatening and fatal meningococcal infections have occurred in patients treated with complement inhibitors. These infections may become rapidly life-threatening or fatal if not recognized and treated early.
- Complete or update vaccination for meningococcal bacteria (for serogroups A, C, W, Y, and B) at least 2 weeks prior to the first dose of ULTOMIRIS, unless the risks of delaying ULTOMIRIS therapy outweigh the risk of developing a serious infection. Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for vaccinations against meningococcal bacteria in patients receiving a complement inhibitor. See Warnings and Precautions (5.1) for additional guidance on the management of the risk of serious infections caused by meningococcal bacteria.
- Patients receiving ULTOMIRIS are at increased risk for invasive disease caused by Neisseria meningitidis, even if they develop antibodies following vaccination. Monitor patients for early signs and symptoms of serious meningococcal infections and evaluate immediately if infection is suspected.
Because of the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called ULTOMIRIS and SOLIRIS REMS [see Warnings and Precautions (5.2)].
CONTRAINDICATIONS
- Initiation in patients with unresolved serious Neisseria meningitidis infection.
WARNINGS AND PRECAUTIONS
Serious Meningococcal Infections
ULTOMIRIS, a complement inhibitor, increases a patient’s susceptibility to serious, life-threatening, or fatal infections caused by meningococcal bacteria (septicemia and/or meningitis) in any serogroup, including non-groupable strains. Life-threatening and fatal meningococcal infections have occurred in both vaccinated and unvaccinated patients treated with complement inhibitors.
Revaccinate patients in accordance with ACIP recommendations considering the duration of ULTOMIRIS therapy. Note that ACIP recommends an administration schedule in patients receiving complement inhibitors that differs from the administration schedule in the vaccine prescribing information. If urgent ULTOMIRIS therapy is indicated in a patient who is not up to date with meningococcal vaccines according to ACIP recommendations, provide antibacterial drug prophylaxis and administer meningococcal vaccines as soon as possible. Various durations and regimens of antibacterial drug prophylaxis have been considered, but the optimal durations and drug regimens for prophylaxis and their efficacy have not been studied in unvaccinated or vaccinated patients receiving complement inhibitors, including ULTOMIRIS. The benefits and risks of treatment with ULTOMIRIS, as well as those associated with antibacterial drug prophylaxis in unvaccinated or vaccinated patients, must be considered against the known risks for serious infections caused by Neisseria meningitidis.
Vaccination does not eliminate the risk of serious meningococcal infections, despite development of antibodies following vaccination.
Closely monitor patients for early signs and symptoms of meningococcal infection and evaluate patients immediately if infection is suspected. Inform patients of these signs and symptoms and instruct patients to seek immediate medical care if they occur. Promptly treat known infections. Meningococcal infection may become rapidly life-threatening or fatal if not recognized and treated early. Consider interruption of ULTOMIRIS in patients who are undergoing treatment for serious meningococcal infection depending on the risks of interrupting treatment in the disease being treated.
ULTOMIRIS and SOLIRIS REMS
Due to the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program called ULTOMIRIS and SOLIRIS REMS.
Prescribers must enroll in the REMS, counsel patients about the risk of serious meningococcal infection, provide patients with the REMS educational materials, assess patient vaccination status for meningococcal vaccines (against serogroups A, C, W, Y, and B) and vaccinate if needed according to current ACIP recommendations two weeks prior to the first dose of ULTOMIRIS. Antibacterial drug prophylaxis must be prescribed if treatment must be started urgently, and the patient is not up to date with both meningococcal vaccines according to current ACIP recommendations at least two weeks prior to the first dose of ULTOMIRIS. Patients must receive counseling about the need to receive meningococcal vaccines and to take antibiotics as directed, signs and symptoms of meningococcal infection, and be instructed to carry the Patient Safety Card at all times during and for 8 months following ULTOMIRIS treatment.
Further information is available at www.UltSolREMS.com or 1-888-765-4747.
Other Infections
Serious infections with Neisseria species (other than Neisseria meningitidis), including disseminated gonococcal infections, have been reported.
ULTOMIRIS blocks terminal complement activation; therefore, patients may have increased susceptibility to infections, especially with encapsulated bacteria, such as infections caused by Neisseria meningitidis but also Streptococcus pneumoniae, Haemophilus influenzae, and to a lesser extent, Neisseria gonorrhoeae. Children treated with ULTOMIRIS may be at increased risk of developing serious infections due to Streptococcus pneumoniae and Haemophilus influenzae type b (Hib). Administer vaccinations for the prevention of Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) infections according to ACIP recommendations. Patients receiving ULTOMIRIS are at increased risk for infections due to these organisms, even if they develop antibodies following vaccination.
Monitoring Disease Manifestations after ULTOMIRIS Discontinuation
Treatment Discontinuation for PNH
After discontinuing treatment with ULTOMIRIS, closely monitor for signs and symptoms of hemolysis, identified by elevated LDH along with sudden decrease in PNH clone size or hemoglobin, or re-appearance of symptoms such as fatigue, hemoglobinuria, abdominal pain, shortness of breath (dyspnea), major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction. Monitor any patient who discontinues ULTOMIRIS for at least 16 weeks to detect hemolysis and other reactions. If signs and symptoms of hemolysis occur after discontinuation, including elevated LDH, consider restarting treatment with ULTOMIRIS.
Treatment Discontinuation for aHUS
ULTOMIRIS treatment of aHUS should be a minimum duration of 6 months. Due to heterogeneous nature of aHUS events and patient-specific risk factors, treatment duration beyond the initial 6 months should be individualized. There are no specific data on ULTOMIRIS discontinuation. After discontinuing treatment with ULTOMIRIS, patients should be monitored for clinical symptoms and laboratory signs of TMA complications for at least 12 months. TMA complications post-discontinuation can be identified if any of the following is observed: Clinical symptoms of TMA include changes in mental status, seizures, angina, dyspnea, thrombosis or increasing blood pressure. In addition, at least two of the following laboratory signs observed concurrently and results should be confirmed by a second measurement 28 days apart with no interruption: a decrease in platelet count of 25% or more as compared to either baseline or to peak platelet count during ULTOMIRIS treatment; an increase in serum creatinine of 25% or more as compared to baseline or to nadir during ULTOMIRIS treatment; or, an increase in serum LDH of 25% or more as compared to baseline or to nadir during ULTOMIRIS treatment. If TMA complications occur after discontinuation, consider reinitiation of ULTOMIRIS treatment or appropriate organ-specific supportive measures.
Thromboembolic Event Management
The effect of withdrawal of anticoagulant therapy during treatment with ULTOMIRIS has not been established. Treatment should not alter anticoagulant management.
Infusion-Related Reactions
Administration of ULTOMIRIS may result in systemic infusion-related reactions, including anaphylaxis and hypersensitivity reactions. In clinical trials, infusion-related reactions occurred in approximately 1 to 7% of patients, including lower back pain, abdominal pain, muscle spasms, drop or elevation in blood pressure, rigors, limb discomfort, drug hypersensitivity (allergic reaction), and dysgeusia (bad taste). These reactions did not require discontinuation of ULTOMIRIS. If signs of cardiovascular instability or respiratory compromise occur, interrupt ULTOMIRIS and institute appropriate supportive measures.
ADVERSE REACTIONS
Adverse Reactions for PNH
Adverse reactions reported in ≥10% or more of patients with PNH were upper respiratory tract infection and headache. Serious adverse reactions were reported in 15 (6.8%) patients receiving ULTOMIRIS. The serious adverse reactions in patients treated with ULTOMIRIS included hyperthermia and pyrexia. No serious adverse reaction was reported in more than 1 patient treated with ULTOMIRIS. One fatal case of sepsis was identified in a patient treated with ULTOMIRIS. In clinical studies, clinically relevant adverse reactions in 1% of adult patients include infusion-related reactions.
Adverse reactions reported in ≥10% of pediatric patients treated with ULTOMIRIS who were treatment-naïve vs. Eculizumab-experienced were anemia (20% vs. 25%), abdominal pain (0% vs. 38%), constipation (0% vs. 25%), pyrexia (20% vs. 13%), upper respiratory tract infection (20% vs. 75%), pain in extremity (0% vs. 25%), and headache (20% vs. 25%).
Adverse Reactions for aHUS
Most common adverse reactions in patients with aHUS (incidence ≥20%) were upper respiratory tract infection, diarrhea, nausea, vomiting, headache, hypertension and pyrexia. Serious adverse reactions were reported in 42 (57%) patients with aHUS receiving ULTOMIRIS. The most frequent serious adverse reactions reported in more than 2 patients (2.7%) treated with ULTOMIRIS were hypertension, pneumonia and abdominal pain.
Adverse reactions reported in ≥20% of pediatric patients treated with ULTOMIRIS were diarrhea, constipation, vomiting, pyrexia, upper respiratory tract infection, decreased vitamin D, headache, cough, rash, and hypertension.
Adverse Reactions for gMG
Most common adverse reactions in adult patients with gMG (incidence ≥10%) were diarrhea and upper respiratory tract infection. Serious adverse reactions were reported in 20 (23%) of patients treated with ULTOMIRIS and in 14 (16%) patients receiving placebo. The most frequent serious adverse reactions were infections reported in at least 8 (9%) patients treated with ULTOMIRIS and in 4 (4%) patients treated with placebo. Of these infections, one fatal case of COVID-19 pneumonia was identified in a patient treated with ULTOMIRIS and one case of infection led to discontinuation of ULTOMIRIS.
Adverse Reactions for NMOSD
Most common adverse reactions in adult patients with NMOSD (incidence ≥10%) were COVID-19, headache, back pain, arthralgia, and urinary tract infection. Serious adverse reactions were reported in 8 (13.8%) patients with NMOSD receiving ULTOMIRIS.
DRUG INTERACTIONS
Plasma Exchange, Plasmapheresis, and Intravenous Immunoglobulins
Concomitant use of ULTOMIRIS with plasma exchange (PE), plasmapheresis (PP), or intravenous immunoglobulin (IVIg) treatment can reduce serum ravulizumab concentrations and requires a supplemental dose of ULTOMIRIS.
Neonatal Fc Receptor Blockers
Concomitant use of ULTOMIRIS with neonatal Fc receptor (FcRn) blockers (e.g., efgartigimod) may lower systemic exposures and reduce effectiveness of ULTOMIRIS. Closely monitor for reduced effectiveness of ULTOMIRIS.
USE IN SPECIFIC POPULATIONS
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ULTOMIRIS during pregnancy. Healthcare providers and patients may call 1-833-793-0563 or go to www.UltomirisPregnancyStudy.com to enroll in or to obtain information about the registry.
To report SUSPECTED ADVERSE REACTIONS, contact Alexion Pharmaceuticals, Inc. at 1-844-259-6783 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
INDICATIONS
Paroxysmal Nocturnal Hemoglobinuria (PNH)
ULTOMIRIS is indicated for the treatment of adult and pediatric patients one month of age and older with paroxysmal nocturnal hemoglobinuria (PNH).
Atypical Hemolytic Uremic Syndrome (aHUS)
ULTOMIRIS is indicated for the treatment of adult and pediatric patients one month of age and older with atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy (TMA).
Limitation of Use:
ULTOMIRIS is not indicated for the treatment of patients with Shiga toxin E. coli related hemolytic uremic syndrome (STEC-HUS).
Generalized Myasthenia Gravis (gMG)
ULTOMIRIS is indicated for the treatment of adult patients with generalized myasthenia gravis (gMG) who are anti-acetylcholine receptor (AChR) antibody-positive.
Neuromyelitis Optica Spectrum Disorder (NMOSD)
ULTOMIRIS is indicated for the treatment of adult patients with neuromyelitis optica spectrum disorder (NMOSD) who are anti-aquaporin-4 (AQP4) antibody positive.
Please see full Prescribing Information for ULTOMIRIS, including Boxed WARNING regarding serious and life-threatening or fatal meningococcal infections.
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
ULTOMIRIS, a complement inhibitor, increases the risk of serious infections caused by Neisseria meningitidis [see Warnings and Precautions (5.1)] Life-threatening and fatal meningococcal infections have occurred in patients treated with complement inhibitors. These infections may become rapidly life-threatening or fatal if not recognized and treated early.
- Complete or update vaccination for meningococcal bacteria (for serogroups A, C, W, Y, and B) at least 2 weeks prior to the first dose of ULTOMIRIS, unless the risks of delaying ULTOMIRIS therapy outweigh the risk of developing a serious infection. Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for vaccinations against meningococcal bacteria in patients receiving a complement inhibitor. See Warnings and Precautions (5.1) for additional guidance on the management of the risk of serious infections caused by meningococcal bacteria.
- Patients receiving ULTOMIRIS are at increased risk for invasive disease caused by Neisseria meningitidis, even if they develop antibodies following vaccination. Monitor patients for early signs and symptoms of serious meningococcal infections and evaluate immediately if infection is suspected.
Because of the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called ULTOMIRIS and SOLIRIS REMS [see Warnings and Precautions (5.2)].
CONTRAINDICATIONS
- Initiation in patients with unresolved serious Neisseria meningitidis infection.
WARNINGS AND PRECAUTIONS
Serious Meningococcal Infections
ULTOMIRIS, a complement inhibitor, increases a patient’s susceptibility to serious, life-threatening, or fatal infections caused by meningococcal bacteria (septicemia and/or meningitis) in any serogroup, including non-groupable strains. Life-threatening and fatal meningococcal infections have occurred in both vaccinated and unvaccinated patients treated with complement inhibitors.
Revaccinate patients in accordance with ACIP recommendations considering the duration of ULTOMIRIS therapy. Note that ACIP recommends an administration schedule in patients receiving complement inhibitors that differs from the administration schedule in the vaccine prescribing information. If urgent ULTOMIRIS therapy is indicated in a patient who is not up to date with meningococcal vaccines according to ACIP recommendations, provide antibacterial drug prophylaxis and administer meningococcal vaccines as soon as possible. Various durations and regimens of antibacterial drug prophylaxis have been considered, but the optimal durations and drug regimens for prophylaxis and their efficacy have not been studied in unvaccinated or vaccinated patients receiving complement inhibitors, including ULTOMIRIS. The benefits and risks of treatment with ULTOMIRIS, as well as those associated with antibacterial drug prophylaxis in unvaccinated or vaccinated patients, must be considered against the known risks for serious infections caused by Neisseria meningitidis.
Vaccination does not eliminate the risk of serious meningococcal infections, despite development of antibodies following vaccination.
Closely monitor patients for early signs and symptoms of meningococcal infection and evaluate patients immediately if infection is suspected. Inform patients of these signs and symptoms and instruct patients to seek immediate medical care if they occur. Promptly treat known infections. Meningococcal infection may become rapidly life-threatening or fatal if not recognized and treated early. Consider interruption of ULTOMIRIS in patients who are undergoing treatment for serious meningococcal infection depending on the risks of interrupting treatment in the disease being treated.
ULTOMIRIS and SOLIRIS REMS
Due to the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program called ULTOMIRIS and SOLIRIS REMS.
Prescribers must enroll in the REMS, counsel patients about the risk of serious meningococcal infection, provide patients with the REMS educational materials, assess patient vaccination status for meningococcal vaccines (against serogroups A, C, W, Y, and B) and vaccinate if needed according to current ACIP recommendations two weeks prior to the first dose of ULTOMIRIS. Antibacterial drug prophylaxis must be prescribed if treatment must be started urgently, and the patient is not up to date with both meningococcal vaccines according to current ACIP recommendations at least two weeks prior to the first dose of ULTOMIRIS. Patients must receive counseling about the need to receive meningococcal vaccines and to take antibiotics as directed, signs and symptoms of meningococcal infection, and be instructed to carry the Patient Safety Card at all times during and for 8 months following ULTOMIRIS treatment.
Further information is available at www.UltSolREMS.com or 1-888-765-4747.
Other Infections
Serious infections with Neisseria species (other than Neisseria meningitidis), including disseminated gonococcal infections, have been reported.
ULTOMIRIS blocks terminal complement activation; therefore, patients may have increased susceptibility to infections, especially with encapsulated bacteria, such as infections caused by Neisseria meningitidis but also Streptococcus pneumoniae, Haemophilus influenzae, and to a lesser extent, Neisseria gonorrhoeae. Children treated with ULTOMIRIS may be at increased risk of developing serious infections due to Streptococcus pneumoniae and Haemophilus influenzae type b (Hib). Administer vaccinations for the prevention of Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) infections according to ACIP recommendations. Patients receiving ULTOMIRIS are at increased risk for infections due to these organisms, even if they develop antibodies following vaccination.
Monitoring Disease Manifestations after ULTOMIRIS Discontinuation
After discontinuing treatment with ULTOMIRIS, closely monitor for signs and symptoms of hemolysis, identified by elevated LDH along with sudden decrease in PNH clone size or hemoglobin, or re-appearance of symptoms such as fatigue, hemoglobinuria, abdominal pain, shortness of breath (dyspnea), major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction. Monitor any patient who discontinues ULTOMIRIS for at least 16 weeks to detect hemolysis and other reactions. If signs and symptoms of hemolysis occur after discontinuation, including elevated LDH, consider restarting treatment with ULTOMIRIS.
Thromboembolic Event Management
The effect of withdrawal of anticoagulant therapy during treatment with ULTOMIRIS has not been established. Treatment should not alter anticoagulant management.
Infusion-Related Reactions
Administration of ULTOMIRIS may result in systemic infusion-related reactions, including anaphylaxis and hypersensitivity reactions. In clinical trials, infusion-related reactions occurred in approximately 1 to 7% of patients, including lower back pain, abdominal pain, muscle spasms, drop or elevation in blood pressure, rigors, limb discomfort, drug hypersensitivity (allergic reaction), and dysgeusia (bad taste). These reactions did not require discontinuation of ULTOMIRIS. If signs of cardiovascular instability or respiratory compromise occur, interrupt ULTOMIRIS and institute appropriate supportive measures.
ADVERSE REACTIONS
Adverse reactions reported in ≥10% or more of patients with PNH were upper respiratory tract infection and headache. Serious adverse reactions were reported in 15 (6.8%) patients receiving ULTOMIRIS. The serious adverse reactions in patients treated with ULTOMIRIS included hyperthermia and pyrexia. No serious adverse reaction was reported in more than 1 patient treated with ULTOMIRIS. One fatal case of sepsis was identified in a patient treated with ULTOMIRIS. In clinical studies, clinically relevant adverse reactions in 1% of adult patients include infusion-related reactions.
Adverse reactions reported in ≥10% of pediatric patients treated with ULTOMIRIS who were treatment-naïve vs. Eculizumab-experienced were anemia (20% vs. 25%), abdominal pain (0% vs. 38%), constipation (0% vs. 25%), pyrexia (20% vs. 13%), upper respiratory tract infection (20% vs. 75%), pain in extremity (0% vs. 25%), and headache (20% vs. 25%).
DRUG INTERACTIONS
Plasma Exchange, Plasmapheresis, and Intravenous Immunoglobulins
Concomitant use of ULTOMIRIS with plasma exchange (PE), plasmapheresis (PP), or intravenous immunoglobulin (IVIg) treatment can reduce serum ravulizumab concentrations and requires a supplemental dose of ULTOMIRIS.
Neonatal Fc Receptor Blockers
Concomitant use of ULTOMIRIS with neonatal Fc receptor (FcRn) blockers (e.g., efgartigimod) may lower systemic exposures and reduce effectiveness of ULTOMIRIS. Closely monitor for reduced effectiveness of ULTOMIRIS.
USE IN SPECIFIC POPULATIONS
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ULTOMIRIS during pregnancy. Healthcare providers and patients may call 1-833-793-0563 or go to www.UltomirisPregnancyStudy.com to enroll in or to obtain information about the registry.
INDICATION
ULTOMIRIS is indicated for the treatment of adult and pediatric patients one month of age and older with paroxysmal nocturnal hemoglobinuria (PNH).
To report SUSPECTED ADVERSE REACTIONS, contact Alexion Pharmaceuticals, Inc. at 1-844-259-6783 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Please see full Prescribing Information for ULTOMIRIS, including Boxed WARNING regarding serious and life-threatening or fatal meningococcal infections.
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
ULTOMIRIS, a complement inhibitor, increases the risk of serious infections caused by Neisseria meningitidis [see Warnings and Precautions (5.1)] Life-threatening and fatal meningococcal infections have occurred in patients treated with complement inhibitors. These infections may become rapidly life-threatening or fatal if not recognized and treated early.
- Complete or update vaccination for meningococcal bacteria (for serogroups A, C, W, Y, and B) at least 2 weeks prior to the first dose of ULTOMIRIS, unless the risks of delaying ULTOMIRIS therapy outweigh the risk of developing a serious infection. Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for vaccinations against meningococcal bacteria in patients receiving a complement inhibitor. See Warnings and Precautions (5.1) for additional guidance on the management of the risk of serious infections caused by meningococcal bacteria.
- Patients receiving ULTOMIRIS are at increased risk for invasive disease caused by Neisseria meningitidis, even if they develop antibodies following vaccination. Monitor patients for early signs and symptoms of serious meningococcal infections and evaluate immediately if infection is suspected.
Because of the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called ULTOMIRIS and SOLIRIS REMS [see Warnings and Precautions (5.2)].
CONTRAINDICATIONS
- Initiation in patients with unresolved serious Neisseria meningitidis infection.
WARNINGS AND PRECAUTIONS
Serious Meningococcal Infections
ULTOMIRIS, a complement inhibitor, increases a patient’s susceptibility to serious, life-threatening, or fatal infections caused by meningococcal bacteria (septicemia and/or meningitis) in any serogroup, including non-groupable strains. Life-threatening and fatal meningococcal infections have occurred in both vaccinated and unvaccinated patients treated with complement inhibitors.
Revaccinate patients in accordance with ACIP recommendations considering the duration of ULTOMIRIS therapy. Note that ACIP recommends an administration schedule in patients receiving complement inhibitors that differs from the administration schedule in the vaccine prescribing information. If urgent ULTOMIRIS therapy is indicated in a patient who is not up to date with meningococcal vaccines according to ACIP recommendations, provide antibacterial drug prophylaxis and administer meningococcal vaccines as soon as possible. Various durations and regimens of antibacterial drug prophylaxis have been considered, but the optimal durations and drug regimens for prophylaxis and their efficacy have not been studied in unvaccinated or vaccinated patients receiving complement inhibitors, including ULTOMIRIS. The benefits and risks of treatment with ULTOMIRIS, as well as those associated with antibacterial drug prophylaxis in unvaccinated or vaccinated patients, must be considered against the known risks for serious infections caused by Neisseria meningitidis.
Vaccination does not eliminate the risk of serious meningococcal infections, despite development of antibodies following vaccination.
Closely monitor patients for early signs and symptoms of meningococcal infection and evaluate patients immediately if infection is suspected. Inform patients of these signs and symptoms and instruct patients to seek immediate medical care if they occur. Promptly treat known infections. Meningococcal infection may become rapidly life-threatening or fatal if not recognized and treated early. Consider interruption of ULTOMIRIS in patients who are undergoing treatment for serious meningococcal infection depending on the risks of interrupting treatment in the disease being treated.
ULTOMIRIS and SOLIRIS REMS
Due to the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program called ULTOMIRIS and SOLIRIS REMS.
Prescribers must enroll in the REMS, counsel patients about the risk of serious meningococcal infection, provide patients with the REMS educational materials, assess patient vaccination status for meningococcal vaccines (against serogroups A, C, W, Y, and B) and vaccinate if needed according to current ACIP recommendations two weeks prior to the first dose of ULTOMIRIS. Antibacterial drug prophylaxis must be prescribed if treatment must be started urgently, and the patient is not up to date with both meningococcal vaccines according to current ACIP recommendations at least two weeks prior to the first dose of ULTOMIRIS. Patients must receive counseling about the need to receive meningococcal vaccines and to take antibiotics as directed, signs and symptoms of meningococcal infection, and be instructed to carry the Patient Safety Card at all times during and for 8 months following ULTOMIRIS treatment.
Further information is available at www.UltSolREMS.com or 1-888-765-4747.
Other Infections
Serious infections with Neisseria species (other than Neisseria meningitidis), including disseminated gonococcal infections, have been reported.
ULTOMIRIS blocks terminal complement activation; therefore, patients may have increased susceptibility to infections, especially with encapsulated bacteria, such as infections caused by Neisseria meningitidis but also Streptococcus pneumoniae, Haemophilus influenzae, and to a lesser extent, Neisseria gonorrhoeae. Children treated with ULTOMIRIS may be at increased risk of developing serious infections due to Streptococcus pneumoniae and Haemophilus influenzae type b (Hib). Administer vaccinations for the prevention of Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) infections according to ACIP recommendations. Patients receiving ULTOMIRIS are at increased risk for infections due to these organisms, even if they develop antibodies following vaccination.
Monitoring Disease Manifestations after ULTOMIRIS Discontinuation
ULTOMIRIS treatment of aHUS should be a minimum duration of 6 months. Due to heterogeneous nature of aHUS events and patient-specific risk factors, treatment duration beyond the initial 6 months should be individualized. There are no specific data on ULTOMIRIS discontinuation. After discontinuing treatment with ULTOMIRIS, patients should be monitored for clinical symptoms and laboratory signs of TMA complications for at least 12 months. TMA complications post-discontinuation can be identified if any of the following is observed: Clinical symptoms of TMA include changes in mental status, seizures, angina, dyspnea, thrombosis or increasing blood pressure. In addition, at least two of the following laboratory signs observed concurrently and results should be confirmed by a second measurement 28 days apart with no interruption: a decrease in platelet count of 25% or more as compared to either baseline or to peak platelet count during ULTOMIRIS treatment; an increase in serum creatinine of 25% or more as compared to baseline or to nadir during ULTOMIRIS treatment; or, an increase in serum LDH of 25% or more as compared to baseline or to nadir during ULTOMIRIS treatment. If TMA complications occur after discontinuation, consider reinitiation of ULTOMIRIS treatment or appropriate organ-specific supportive measures.
Thromboembolic Event Management
The effect of withdrawal of anticoagulant therapy during treatment with ULTOMIRIS has not been established. Treatment should not alter anticoagulant management.
Infusion-Related Reactions
Administration of ULTOMIRIS may result in systemic infusion-related reactions, including anaphylaxis and hypersensitivity reactions. In clinical trials, infusion-related reactions occurred in approximately 1 to 7% of patients, including lower back pain, abdominal pain, muscle spasms, drop or elevation in blood pressure, rigors, limb discomfort, drug hypersensitivity (allergic reaction), and dysgeusia (bad taste). These reactions did not require discontinuation of ULTOMIRIS. If signs of cardiovascular instability or respiratory compromise occur, interrupt ULTOMIRIS and institute appropriate supportive measures.
ADVERSE REACTIONS
Most common adverse reactions in patients with aHUS (incidence ≥20%) were upper respiratory tract infection, diarrhea, nausea, vomiting, headache, hypertension and pyrexia. Serious adverse reactions were reported in 42 (57%) patients with aHUS receiving ULTOMIRIS. The most frequent serious adverse reactions reported in more than 2 patients (2.7%) treated with ULTOMIRIS were hypertension, pneumonia and abdominal pain.
Adverse reactions reported in ≥20% of pediatric patients treated with ULTOMIRIS were diarrhea, constipation, vomiting, pyrexia, upper respiratory tract infection, decreased vitamin D, headache, cough, rash, and hypertension.
DRUG INTERACTIONS
Plasma Exchange, Plasmapheresis, and Intravenous Immunoglobulins
Concomitant use of ULTOMIRIS with plasma exchange (PE), plasmapheresis (PP), or intravenous immunoglobulin (IVIg) treatment can reduce serum ravulizumab concentrations and requires a supplemental dose of ULTOMIRIS.
Neonatal Fc Receptor Blockers
Concomitant use of ULTOMIRIS with neonatal Fc receptor (FcRn) blockers (e.g., efgartigimod) may lower systemic exposures and reduce effectiveness of ULTOMIRIS. Closely monitor for reduced effectiveness of ULTOMIRIS.
USE IN SPECIFIC POPULATIONS
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ULTOMIRIS during pregnancy. Healthcare providers and patients may call 1-833-793-0563 or go to www.UltomirisPregnancyStudy.com to enroll in or to obtain information about the registry.
INDICATION
ULTOMIRIS is indicated for the treatment of adult and pediatric patients one month of age and older with atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy (TMA).
Limitation of Use:
ULTOMIRIS is not indicated for the treatment of patients with Shiga toxin E. coli related hemolytic uremic syndrome (STEC‑HUS).
To report SUSPECTED ADVERSE REACTIONS, contact Alexion Pharmaceuticals, Inc. at 1-844-259-6783 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Please see full Prescribing Information for ULTOMIRIS, including Boxed WARNING regarding serious and life-threatening or fatal meningococcal infections.